



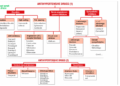
Quality Assurance Department
“Quality” is the excellence that is better than a minimum standard.
“Assurance” means confidence or certainty in one’s work or abilities.
Understanding these two terms, we simply can articulate that quality assurance means assuring one’s work, ability or quality of the product and hence approved. According to WHO-“Quality assurance” is a wide-ranging concept covering all matters that individually or collectively influence the quality of a product.
| Quality Assurance (QA)= Good manufacturing practice(GMP) + Quality control (QC) |
Quality assurance is the sum of organized arrangement and all planned and systematic actions necessary to provide adequate confidence that the product will satisfy the requirements for quality and can be approved for intended use. Quality control is a part of GMP and GMP is a part of QA thus, they are an interrelated system.
Responsibilities of Quality Assurance Department:
Aforementioned, QA is a wide ranging concept that covers all the matters; it plays one of the crucial roles in an organization.
1. Implementation of GMP – GMPrefers to the Good Manufacturing Practice Regulations promulgated by the US Food and Drug Administration under the authority of the Federal Food, Drug, and Cosmetic Act. GMP is the aspect of quality assurance which ensures that medicinal products are produced and controlled to the quality standards appropriate to their intended use as required by the product specification. GMP regulations require a quality approach to minimize or eliminate instances of contamination and mix-ups. This in turn, protects the consumer from purchasing a product which is not effective or even dangerous. GMP regulations address issues including record keeping, personnel qualifications, sanitation, cleanliness, equipment verification, process validation, and focuses on legal components, covering responsibilities for distribution, manufacturing and packaging, and responses to product defects and complaints. Thus, it is the responsibility of QA to concentrate whether the whole organization is working according to GMP guidelines or not.
2. Documentation and control – Documentation generally provides the justification that the work has been done. Documentation access following information:
-
- Provides detail information of the procedures and specifications for all the materials and methods.
- Provides an evidence of performed work or analysis and helps in traceability.
- Provides records and audit trait that will permit investigation.
- Provides all the data which is needed for future analysis or review.
Role played by QA in Documentation:
-
- Classification of documents
- Coding of documents
- Maintenance of the master copies of every documents
- Distributing the controlled documents to authorized person when required
- Periodic review of documents
- Updating all the new or additional information in/about a document and preventing the unintended use of obsolete documents
Documents maintained by Quality Assurance Department
- Standard Operating Procedure (SOPs): SOP is the detail instructions which describes regular occurring procedures appropriate to quality operations. It is based on what is done and what is written and also gives an answer for ‘how to’ perform a task. Most importantly the main purpose to design SOP is to bring uniformity in performance. Each institute functions differently according to varying circumstances, for which they have their own set of procedures to perform certain works. Specific code is given to each SOP and information is updated. In case of any new document or new procedure to undertake certain works, then concerned personnel are trained and after clear understanding the effective date is given for implementation of that standard procedure. Review of SOP is done on periodic basis and if necessary changes are recommended. Controlled copy which is distributed within the organization has a stamp of CONTROLLED COPY, uncontrolled copy which is generally given to bodies outside the organization is stamped as UNCONTROLLED COPY.
- Site master file: Site master file is a document prepared by the manufacturer which provides complete and factual information about the organization. It is imperative as it imparts entire description about an organization. Information mentioned below is given in site master file:
- Name and address of the organization
- Description plant layout
- Types of products manufactured
- Manpower engaged
- Premises and equipment
- Documentation
- Production in brief
- Quality control system,
- Distribution, complaints and product recall
- Inspection, etc.
- Batch Manufacturing Record: – Batch manufacturing record is preparedto provide information relating to the manufacturing process undertaken for a particular batch. They are numbered individually and dated and signed when issued. Main function is to trace the complete cycle of a batch. BMR consist of following
- Name of the product and its batch size
- Equipment used
- API and excipients used along with its quantity
- Manufacturing steps starting from dispensing goods from warehouse till the bulk product formulation
- Parameters recorded
- Result of analysis
- Name and signature of supervisor
- Name and signature of operators
- Yield value
- Batch Packaging Record (BPR):BPR is prepared to provide all the information relating to packaging of a particular batch. Before any processing begins the work area and the equipment used are checked carefully and ensured that they are clean. BPR consists of following information:
- Name of the product
- Date and time of packaging
- Name of the responsible person carrying out the packaging
- Record of check of product
- Detail about packaging operations
- Additional over printing is done
- Manufacture date and expiry date along with batch number is mentioned.
- Record of material destruction: During manufacturing some of the drugs or some raw materials are rejected. In such a case these rejected materials should be destroyed. Record should be maintained to write down which material in how much amount is being destroyed. Reason for its destruction should also be mentioned clearly and process of destruction should be stated.
- Market complain file: After distribution of products in the market, at any case if a complaint has been reported then it should be addressed. The live defected sample is sent along with the detail report of complaint. QA inspects and solves the problem aroused. After the problem has been solved QA also arrange meeting and training to avoid such problems later. Defects are generally categorized in minor defects and major defects. Problem is solved within a day or as soon as possible.
- New document proposal form:This is the initial stage to propose a new document. When there isunavailability of any new document, this form should be filled up stating name of the document and why this document is required. The documents which can be affected by the formation of this document should also be clearly mentioned so that it won’t be problematic afterwards.
- Document change request form: When the old documents are revised periodically, some changes on the documents can be necessary. Accordingly, this form should be filled up and changes should be done. After the modification training should be given to all the concerned personnel or departments as a whole so that people related to that particular work will be updated. The reason for change should be clearly stated. Generally, documents are reviewed once in 3 years. The record of each batch is generally kept for 5 years after its expiry which helps in data generation. After the new documents have been prepared then it is compulsory to collect all the old documents and distribute the new ones to avoid confusion and miscommunication.
3. Monitoring: One of the major responsibilities is to monitor all the departments within the organization. The main aim of monitoring to avoid mix-up, contamination and cross contamination. Not only department as a whole but QA also monitors individual officers and workers. Monitoring is done according to the guidelines given by GMP. Regions of QA monitoring:
-
- Personnel:
- Are they wearing protective measures during work?
- Are they performing their assigned jobs and interfering in unnecessary tasks?
- Premises: Cleanliness, lights, electricity, wall paints, drainage etc. are checked whether it is GMP friendly or not. And also scrutinize whether the processing rooms are cleaned and equipped according to the requisite. For example, temperature and humidity air flow of the room.
- Equipment: Whether the machines used are clean or not.
- Impact of machine on the product
- Material of the machine (mainly requires a stainless steel of 316 or 304 or Grade)
- Calibration of instrument before using
- Each and every step of processing.
- Personnel:
4. External audit and Self inspection: Systematic and independent examination is done to determine whether the activities or results comply with the standard or not and whether these standards are implemented effectively and is helpful in eliciting product quality. Training is conveyed under ISO terms. If any NC has been allocated during main audit then during follow audit in 3-4 months the mistake should be corrected and if done as according to the requirement the person can request to close NC. Internal observation report is filled during the audit. This is very important for timely correction of potential problems.
External audit is generally done by DDA and the implementation of GMP and ISO guidelines are checked.USA audit or international audit is not on regular visit but when contract has been made with foreign countries then people from international levels visit to inspect how the things are going on.
Self-inspections or internal audit are done within the organization to identity problems internally and correct them prior to external audit. Allocating and tracing the problems or non-conformities of the organization by people working within the organization can be little bit difficult but it is very important as it helps the organization to work in a good pace and improve the product and process quality.
5. Corrective Action and Preventive Action (CAPA) and Non-Conformities (NC):Mistakes can arise while performing any task. It’s very necessary that we learn from our mistakes and try to minimize it when we perform the task again. First of all, the cause of mistake is analyzed which can be termed as root cause analysis, then corrective action is taken in order to correct the error and to be confident that such errors won’t occur in future preventive action is taken.
Non-conformities mean complaints during the internal audit. Incident deviation also lies in non-conformities.
CAPA and NC are recorded during audit and it should be closed once the problem is solved and corrective action and preventive action is implemented.
6. Deviation and change control: When a departure occurs from established procedure or accepted standard then it is called deviation. They are of two types:
- Planned deviation and
- Unplanned deviation
Planned deviation means that we can know fluctuation can arise before they occur. Example: If an instrument is not calibrated before use then we can assume that changes in data will arise. On the other hand, unplanned deviations also known as incidental deviations which means failure of the procedure or material occurs and changes arises suddenly.
Change control can be defined as a planned document to bring changes in the document/procedure/equipment permanently. If the procedure where deviation occurred is used again and again then it becomes change control.
Deviation is always temporary while change control is always permanent.
7. Protocol and format: Protocol is an official procedure to carry out some activities. Basically it is documentation of how things are done.
Some of the protocols available are:
- QC protocols
- Validation protocols
Format is the pattern or way in which things are written and helps in mentioning all the necessary details.
8. Dispensing raw material slip: QA monitors whether the warehouse is dispensing the right material in right amount or not. The approved goods are provided with dispensing slip which contains the name of a material its expiry date and weight dispensed. Then it further goes to the production and packaging department.
9. Quality assurance system manual: Quality assurance system manual is a manual which provides detail information about how the Quality Assurance Department works.
10. Annual product review: All the products manufactured in one whole year should be reviewed to see in which trend the production and marketing is going on. Here the products are assayed and it helps in setting the range for each parameters. For example: pH of the product is checked.
11. Validation and qualification: Validation is documented evidence that provides a high degree of assurance and conformation that the process used is capable of consistently producing the same result that meet the established specification.
-
- Equipment Validation
- System Validation
- Cleaning validation
- Process validation
- Method validation
Qualification of a new machine is done. All system and equipment should be qualified. It gives the assurance and confidence that our product is of quality.
-
- Design qualification(DQ):
- Operational qualification(OQ):
- Performance qualification (PQ):
- Requalification(RQ):
- Factory Acceptance test (FAT):
- Site Acceptance test (SAT):
12. Organizing Meeting: QA organizes meeting when required for discussion of different matters and conclude one good decision and also makes sure that the decision is well implemented.
13. Training: Training for all human resources is very important to make them work according the terms and conditions of an organizations. New staffs are given training for basic knowledge and also to make sure that are well informed about job they are assigned.QA also arranges for training programs when anybody from other departments request for guidance. Instruction about new documents and procedures help to keep all the officers, staffs and workers updated. After the training, questionnaires can be distributed and checked how much did the attendants understand. If necessary training can be re-organized so that all the attendants are clear about the work or changes made. When a new machine arrives then the company itself sends a person to give instruction for proper processing and handling of an instrument.
14. Control sample: After preparation of each batch, some products are kept as control sample (retention sample) in control sample room. This is important to check the stability and state of the product after marketing. At any case if any defect or complaint has been reported then it can be cross checked with a control sample which also acts as an evidence.
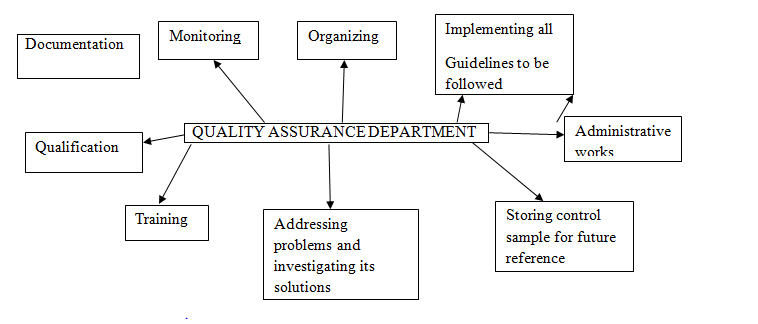
Figure: Areas mostly focused by Quality Assurance Department.
No product or process can be of quality until and unless approved and checked by Quality Assurance. Hence, Quality Assurance Department has pivotal role in maintaining product of good quality which effective and safe.
- How Medicine Quality Affects Antimicrobial Resistance and the Medicines Supply Chain
- Standard Treatment Protocol of Emergency Health Service Package
- Quality Control Tests of Tablet





{kind=link}
Discussion about this post