



Clinical Trials of Drugs
Human testing of new drugs in the United States requires approval by institutional committees that monitor the ethical (informed consent, patient safety) and scientific aspects (study design, statistical power) of the proposed tests. Such testing also requires the prior approval by the FDA of an Investigational New Drug Exemption application (IND), which is submitted by the manufacturer to the FDA. The IND includes all the preclinical data collected up to the time of submission and the detailed proposal for clinical trials. The major clinical testing process is usually divided into 3 phases that are carried out to provide information for a New Drug Application (NDA). The NDA includes all the results of preclinical and clinical testing and constitutes the request for FDA approval of general marketing of the new agent for prescription use. A fourth phase of study (the surveillance phase) follows NDA approval. In particularly lethal conditions, the FDA may permit carefully monitored treatment of patients before phases 2 and 3 are completed.
Phase 1
A phase 1 trial consists of careful evaluation of the dose-response relationship and the pharmacokinetics of the new drug in a small number of normal human volunteers (eg, 20–100). An exception is the phase 1 trials of cancer chemotherapeutic agents and other highly toxic drugs; these are carried out by administering the agents to volunteer patients with the target disease. In phase 1 studies, the acute effects of the agent are studied over a broad range of dosages, starting with one that produces no detectable effect and progressing to one that produces either a significant physiologic response or a very minor toxic effect.
Phase 2
A phase 2 trial involves evaluation of a drug in a moderate number of sick patients (eg, 100–200) with the target disease. A placebo or positive control drug is included in a single-blind or double-blind design. The study is carried out under very carefully controlled conditions, and patients are closely monitored, often in a hospital research ward. The goal is to determine whether the agent has the desired efficacy (ie, produces adequate therapeutic response) at doses that are tolerated by sick patients. Detailed data are collected regarding the pharmacokinetics and pharmacodynamics of the drug in this patient population.
Phase 3
A phase 3 trial usually involves many patients (eg, 1000–6000 or more, in many centers) and many clinicians who are using the drug in the manner proposed for its ultimate general use (eg, in outpatients). Such studies usually include placebo and positive controls in a double-blind crossover design. The goals are to explore further, under the conditions of the proposed clinical use, the spectrum of beneficial actions of the new drug, to compare it with placebo (negative control) and older therapy (positive control), and to discover toxicities, if any, that occur so infrequently as to be undetectable in phase 2 studies. Very large amounts of data are collected and these studies are usually very expensive. Unfortunately, relatively few phase 3 trials include the current standard of care as a positive control.If the drug successfully completes phase 3, an NDA is submitted to the FDA. If the NDA is approved, the drug can be marketed and phase 4 begins.
Phase 4
Phase 4 represents the postmarketing surveillance phase of evaluation, in which it is hoped that toxicities that occur very infrequently will be detected and reported early enough to prevent major therapeutic disasters. Manufacturers are required to inform the FDA at regular intervals of all reported untoward drug reactions. Unlike the first 3 phases, phase 4 has not been rigidly regulated by the FDA in the past. Because so many drugs have been found to be unacceptably toxic only after they have been marketed, there is considerable current interest in making phase 4 surveillance more consistent, effective, and informative.
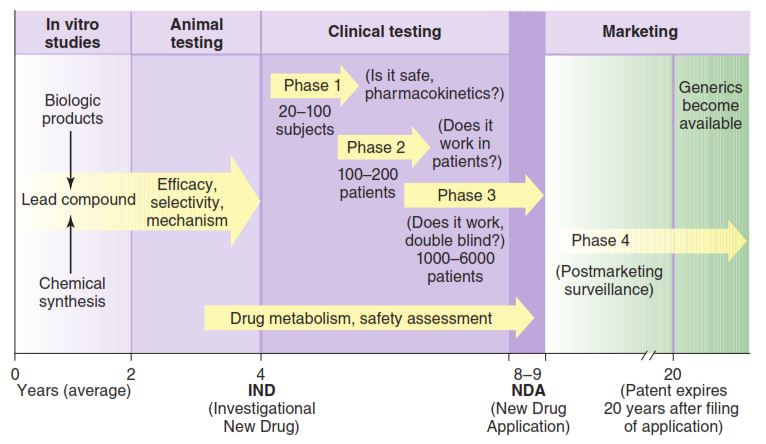
be different for drugs used in life-threatening diseases. (Reproduced, with permission, from Katzung BG, editor: Basic & Clinical Pharmacology12th ed.)







{kind=link}
Discussion about this post